Forschung
Elektronische Struktur, Dynamik und Dichtefunktionaltheorie
Die Forschung unserer Arbeitsgruppe steht im Grenzbereich zwischen Molekülphysik und Festkörperphysik.
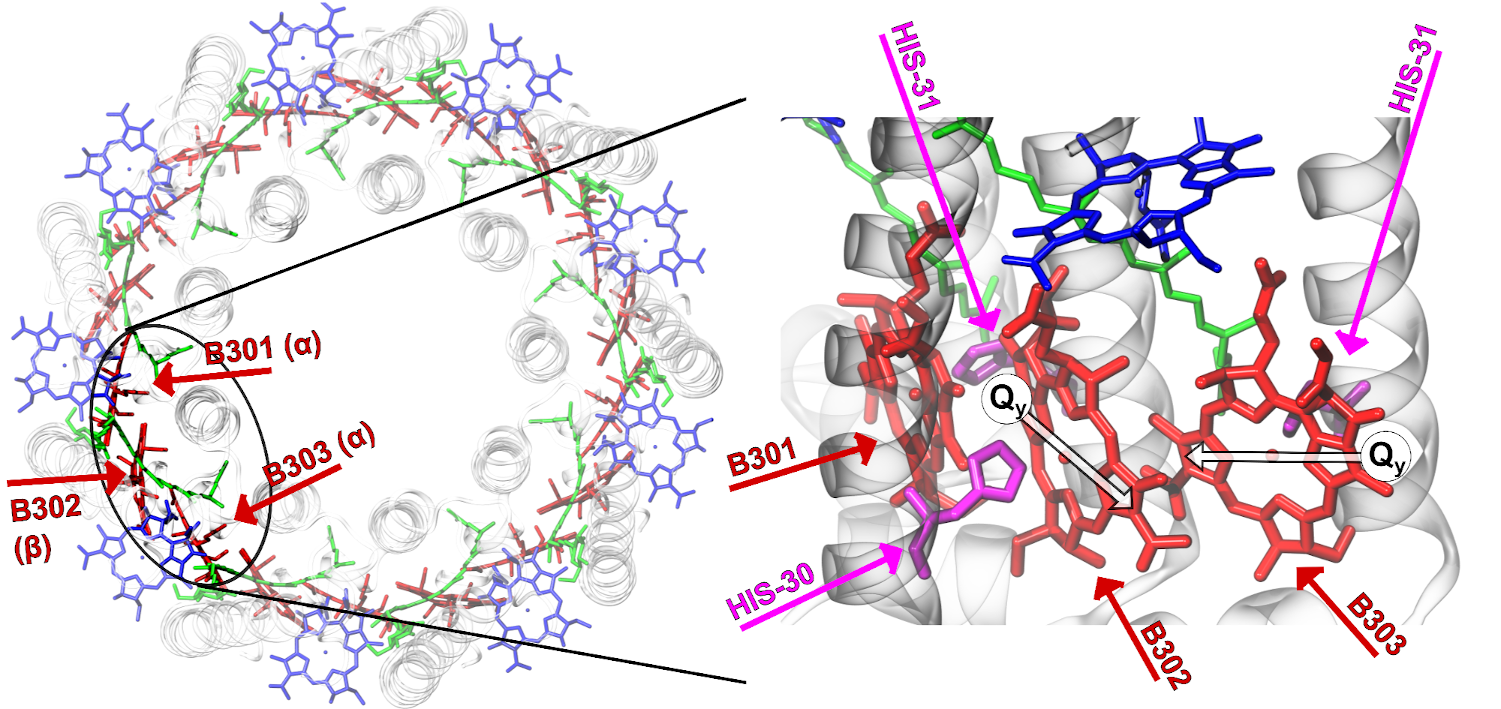
Molekulare Systeme interessieren in unserer Arbeitsgruppe vor allem in Hinblick auf ihre optischen Eigenschaften und Fragen der molekulare Elektronik. Wir möchten z.B. die Wechselwirkung molekularer Halbleiter oder lichtsammelnder Moleküle mit elektrischen Feldern, z.B. bei optischer Anregung, quantitativ berechnen und den Zusammenhang zwischen molekularer Struktur, Anregungen und (auch nichtlinearen) optischen Effekten verstehen. Das Fernziel dabei ist eine voll dynamische "first principles" Beschreibung des Systems aus Kernen, Elektronen und externen Feldern. Ein besonderer Schwerpunkt unserer Forschung liegt auf dem Verständnis von Ladungs- und Energietransfer, z.B. bei Ladungstransferanregungen oder elektrischem Transport durch Molekülketten, oder Energietransfer in Lichtsammelsystemen. Die Vorhersage von Ionisationsenergien und der Zusammenhang zwischen experimentell gemessenen Ionisierungspotentialen und Photoemissionsintensitäten einerseits, und berechneten effektiven Eigenwerten, Orbitalen, und korrelierten Wellenfunktionen andererseits, ist ein weiteres aktives Forschungsfeld unserer Gruppe. Motiviert wird es durch die Hoffnung, durch besseres Verständnis molekularer elektronischer Prozesse einen Beitrag zur Weiterentwicklung von z.B. organischen Solarzellen zu leisten.
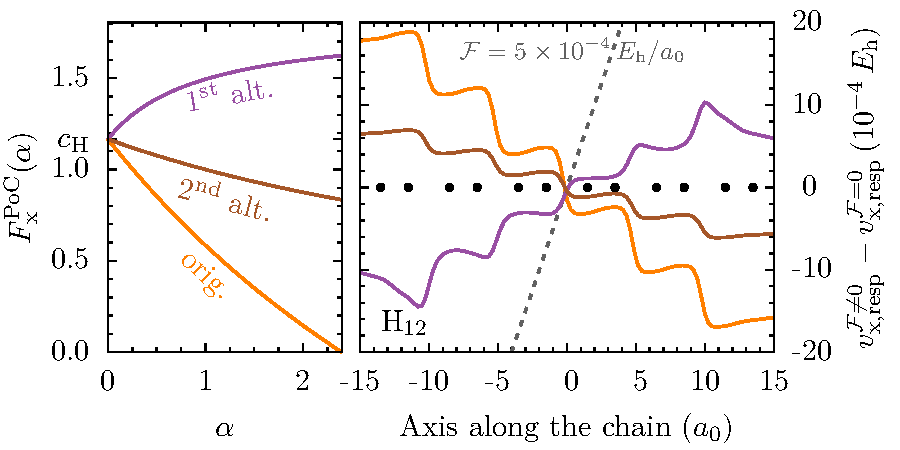
Das zentrale Theorem der Dichtefunktionaltheorie besagt, dass die Grundzustandsdichte alleine ausreicht, um im Prinzip exakt die Eigenschaften eines Vielteilchensystems zu berechnen. Die mit enormem Aufwand verbundene Bestimmung der quantenmechanischen Vielteilchenwellenfunktion wird dabei überflüssig. In der Praxis hängen Genauigkeit und Vorhersagekraft von Dichtefunktionalrechnungen entscheidend von den für das Austausch-Korrelationsfunktional gemachten Näherungen ab. Die Entwicklung besserer Näherungen für dieses Funktional unter expliziter Verwendung der Kohn-Sham Orbitale ("Orbitalfunktionale") ist ein Schwerpunkt unserer Forschung. Die Theorie des "Optimized Effective Potential" spielt dabei eine wichtige Rolle. Unsere Entwicklungen sollen insbesondere auch eine verbesserte Beschreibung zeitabhängiger Probleme ermöglichen, denn die zeitabhängige Dichtefunktionaltheorie ist einer der wenigen Zugänge zur parameter- und modellfreien Berechnung starker, nichtlinearer und nichtperturbativer Anregungen.
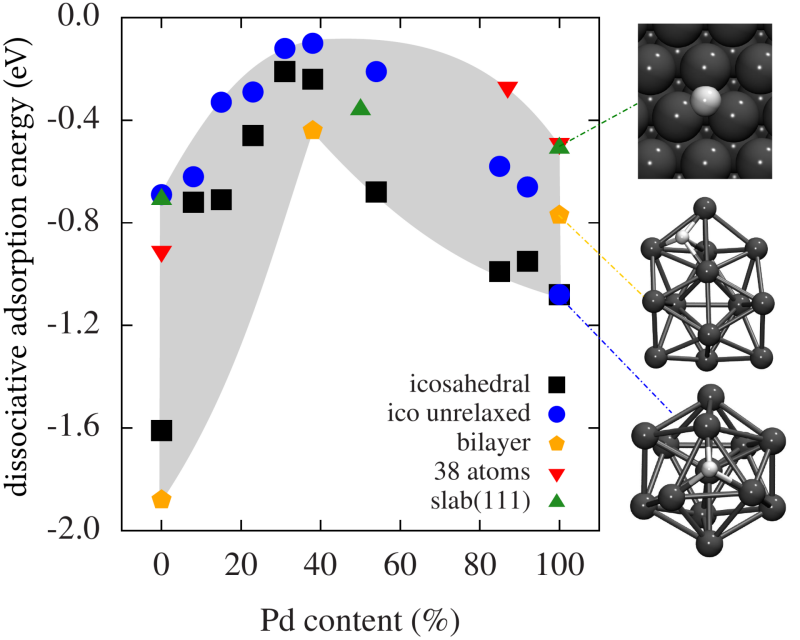
In der Physik der kondensierten Materie stellen Cluster das Bindeglied zwischen der Atom- und Molekülphysik einerseits und der Festkörperphysik andererseits dar. Unter Clustern versteht man Konglomerate aus einigen wenigen bis zu vielen tausend Atomen. Aufgrund ihrer endlichen Größe unterscheiden sich Cluster häufig deutlich in ihren strukturellen und elektronischen Eigenschaften vom kristallinen Festkörper. Daher sind sie sowohl unter fundamentalen Gesichtspunkten von Interesse (Wie wächst Materie vom Atom zum Festkörper? Welche Effekte dominieren das Wachstum? Ab wievielen Atomen stellen sich die Eigenschaften des bulk-Materials ein? Welche optischen Eigenschaften haben Cluster?) als auch unter technologischen (Cluster spielen eine zentrale Rolle in vielen katalytischen Prozessen, sie finden Verwendung in der Nanoelektronik und sind Bausteine für neue Materialien).
Presseerklärungen und Hinweise:
- Emil-Warburg-Preis für Dr. Thomas Körzdörfer: Molekulare Halbleiter
- Blick in die Forschung: Wegweisend für leistungsstarke Solarzellen
- Blick in die Forschung: Elektronen sichtbar gemacht
- Preis der Stadt Bayreuth für Dr. Linn Leppert: Nanolegierungen
- Promotionspreis des GRK 1640 für Dr. Andreas Karolewski
- Focus-Workshop zur Photoemissions-Spektroskopie
- Emil-Warburg-Preis für Dr. Matthias Dauth: "ab initio"-Berechnung der Photoemission